

Anyways the reason why I'm telling you this isn't because I wanted to talk about the project I was doing, (I actually didn't end up finding a lot of genetic differentiation between the two populations), or because I'm warning you of the pitfalls of using aln vs mem, but rather because I think BWA is a really cool concept. BWA and other read mapping programs like it rely on different variations of the same algorithm and represent such a ubiquitous first step in a lot of bioinformatics pipelines. Unless you are a mathematician, most people just run the command without really thinking about what its actually doing in a lot of depth.
BWA - What is it?
BWA stands for Burrows Wheeler Alignment. Its based on an algorithm invented by two guys, Michael Burrows and David Wheeler, called Burrows Wheeler Transform (BWT), that was originally used as a compression tool for text files. It kind of faded into relative obscurity until it was picked up by geneticists in the 2010's who realized that it worked great for DNA sequencing files. In fact almost every read mapping alignment tool for genetics uses the BWT algorithm in some way or another. BWA just happens to be (at the time of writing this) one of the most popular.
It works like this: you take a piece of text, (lets use the example below of the word banana), add a $ symbol to delineate the end, and create columns of each rotation of the letters. So first you would move the letter "b" to the end and shift the "a" up, then in the next row move "a" to the end and shift the "n" up, and so forth.

Fig 1
After you have every combination of the text, you can simplify it by taking just the last column (highlighted in red in Figure 1). For mathematical reasons, when you do this it tends to make the same letters bunch together. You then add a number before each group repeated letters designating how many repeats there are.

Now in this specific instance actual size of the text did not change, so the file containing the text 'banana' would not be compressed. But you can imagine how bunching repeats in this way can be useful for compressing a 50 GB genomic sequencing file of just A's, T's, C's, and G's. But we can take this a step further.
Map those reads!
See the reason why BWA is such a ubiquitous first step in a lot of pipeline's isn't just for its usefulness at compressing files, its also great for aligning newly sequenced genomes to a reference, also known as read mapping.
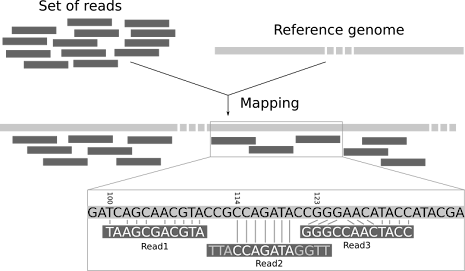
Read mapping basically allows you to take your fragmented genome sequence pieces and match them to the areas of best fit on a reference so you can reorganize them correctly.
The way BWA does this is a bit counter intuitive and difficult to explain in paragraph form but bear with me. First we must create a matrix with four columns. The first column is just a number from 0 to however many characters there are. We label this as i. Then go back to the columns of rotations from before. The second and third columns of our matrix are the first and last columns of letters (highlighted below in red boxes). We label these as "First" and "Last." The fourth column is the most confusing. Using the three columns we have just created in our matrix, we then go row by row and look to see where does the character in the "Last" column, appear primarily in the "First" column and write down the corresponding number from i. We call this L2F(i). Again here's what that looks like using 'banana:'

Now what seems like utterly useless playing around with letters is actually genius. Basically in a very roundabout way, what we've done is make it easier to match where a piece of text might fit best to the reference. For a more detailed explanation on how this occurs see this fantastic youtube tutorial by Niema Moshiri. It involves limiting the range in which a pattern appears using the L2F(i) column. Luckily, we don't have to think about this too hard since we can have a computer do it for us. Again, you can see how helpful this could be with large DNA files and why I get a little bit geeked out when I talk about it. With BWA done and your short read sequences mapped to a reference in their correct order, the next steps of actually analyzing them has become much easier.
No comments:
Post a Comment